| Cytochrome P450 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
.png) Structure of lanosterol 14α-demethylase (
CYP51) | |||||||||
| Identifiers | |||||||||
| Symbol | p450 | ||||||||
| Pfam | PF00067 | ||||||||
| InterPro | IPR001128 | ||||||||
| PROSITE | PDOC00081 | ||||||||
| SCOP2 | 2cpp / SCOPe / SUPFAM | ||||||||
| OPM superfamily | 39 | ||||||||
| OPM protein | 2bdm | ||||||||
| CDD | cd00302 | ||||||||
| Membranome | 265 | ||||||||
| |||||||||
Cytochromes P450 (P450s or CYPs) are a superfamily of enzymes containing heme as a cofactor that mostly, but not exclusively, function as monooxygenases. [1] However, they are not omnipresent; for example, they have not been found in Escherichia coli. [2] In mammals, these enzymes oxidize steroids, fatty acids, xenobiotics, and participate in many biosyntheses. [1] By hydroxylation, CYP450 enzymes convert xenobiotics into hydrophilic derivatives, which are more readily excreted.
P450s are, in general, the terminal oxidase enzymes in electron transfer chains, broadly categorized as P450-containing systems. The term "P450" is derived from the spectrophotometric peak at the wavelength of the absorption maximum of the enzyme (450 nm) when it is in the reduced state and complexed with carbon monoxide. Most P450s require a protein partner to deliver one or more electrons to reduce the iron (and eventually molecular oxygen).
Nomenclature
Genes encoding P450 enzymes, and the enzymes themselves, are designated with the root symbol CYP for the superfamily, followed by a number indicating the gene family, a capital letter indicating the subfamily, and another numeral for the individual gene. The convention is to italicize the name when referring to the gene. For example, CYP2E1 is the gene that encodes the enzyme CYP2E1—one of the enzymes involved in paracetamol (acetaminophen) metabolism. The CYP nomenclature is the official naming convention, although occasionally CYP450 or CYP450 is used synonymously. These names should never be used as according to the nomenclature convention (as they denote a P450 in family number 450). However, some gene or enzyme names for P450s are also referred to by historical names (e.g. P450BM3 for CYP102A1) or functional names, denoting the catalytic activity and the name of the compound used as substrate. Examples include CYP5A1, thromboxane A2 synthase, abbreviated to TBXAS1 (ThromBoXane A2 Synthase 1), and CYP51A1, lanosterol 14-α-demethylase, sometimes unofficially abbreviated to LDM according to its substrate (Lanosterol) and activity (DeMethylation). [3]
The current nomenclature guidelines suggest that members of new CYP families share at least 40% amino-acid identity, while members of subfamilies must share at least 55% amino-acid identity. Nomenclature committees assign and track both base gene names ( Cytochrome P450 Homepage Archived 2010-06-27 at the Wayback Machine) and allele names ( CYP Allele Nomenclature Committee). [4] [5]
Classification
Based on the nature of the electron transfer proteins, P450s can be classified into several groups: [6]
- Microsomal P450 systems
- in which electrons are transferred from NADPH via cytochrome P450 reductase (variously CPR, POR, or CYPOR). Cytochrome b5 (cyb5) can also contribute reducing power to this system after being reduced by cytochrome b5 reductase (CYB5R).
- Mitochondrial P450 systems
- which employ adrenodoxin reductase and adrenodoxin to transfer electrons from NADPH to P450.
- Bacterial P450 systems
- which employ a ferredoxin reductase and a ferredoxin to transfer electrons to P450.
- CYB5R/cyb5/P450 systems
- in which both electrons required by the CYP come from cytochrome b5.
- FMN/Fd/P450 systems
- originally found in Rhodococcus species, in which a FMN-domain-containing reductase is fused to the CYP.
- P450 only systems
- which do not require external reducing power. Notable ones include thromboxane synthase (CYP5), prostacyclin synthase (CYP8), and CYP74A ( allene oxide synthase).
The most common reaction catalyzed by cytochromes P450 is a monooxygenase reaction, e.g., insertion of one atom of oxygen into the aliphatic position of an organic substrate (RH), while the other oxygen atom is reduced to water:
Related hydroxylation enzymes
Many hydroxylation reactions (insertion of hydroxyl groups) use CYP enzymes, but many other hydroxylases exist. Alpha-ketoglutarate-dependent hydroxylases also rely on an Fe=O intermediate but lack hemes. Methane monooxygenase, which converts methane to methanol, are non-heme iron-and iron-copper-based enzymes. [7]
Mechanism

Structure
The active site of cytochrome P450 contains a heme-iron center. The iron is tethered to the protein via a cysteine thiolate ligand. This cysteine and several flanking residues are highly conserved in known P450s, and have the formal PROSITE signature consensus pattern [FW] - [SGNH] - x - [GD] - {F} - [RKHPT] - {P} - C - [LIVMFAP] - [GAD]. [8] In general, the P450 catalytic cycle proceeds as follows:
Catalytic cycle
- Substrate binds in proximity to the heme group, on the side opposite to the axial thiolate. Substrate binding induces a change in the conformation of the active site, often displacing a water molecule from the distal axial coordination position of the heme iron, [9] and changing the state of the heme iron from low-spin to high-spin. [10]
- Substrate binding induces electron transfer from NAD(P)H via cytochrome P450 reductase or another associated reductase, [11] converting Fe(III) to Fe(II).
- Molecular oxygen binds to the resulting ferrous heme center at the distal axial coordination position, initially giving a dioxygen adduct similar to oxy-myoglobin.
- A second electron is transferred, from either cytochrome P450 reductase, ferredoxins, or cytochrome b5, reducing the Fe-O2 adduct to give a short-lived peroxo state.
- The peroxo group formed in step 4 is rapidly protonated twice, releasing one molecule of water and forming the highly reactive species referred to as P450 Compound 1 (or just Compound I). This highly reactive intermediate was isolated in 2010, [12] P450 Compound 1 is an iron(IV) oxo (or ferryl) species with an additional oxidizing equivalent delocalized over the porphyrin and thiolate ligands. Evidence for the alternative perferryl iron(V)-oxo [9] is lacking. [12]
- Depending on the substrate and enzyme involved, P450 enzymes can catalyze any of a wide variety of reactions. A hypothetical hydroxylation is illustrated. After the hydroxylated product has been released from the active site, the enzyme returns to its original state, with a water molecule returning to occupy the distal coordination position of the iron nucleus.
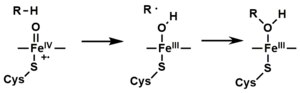
- An alternative route for mono-oxygenation is via the "peroxide shunt" (path "S" in figure). This pathway entails oxidation of the ferric-substrate complex with oxygen-atom donors such as peroxides and hypochlorites. [13] A hypothetical peroxide "XOOH" is shown in the diagram.
Mechanistic details, including the oxygen rebound mechanism, have been investigated with synthetic analogues, consisting of iron oxo heme complexes. [14]
Spectroscopy
Binding of substrate is reflected in the spectral properties of the enzyme, with an increase in absorbance at 390 nm and a decrease at 420 nm. This can be measured by difference spectroscopies and is referred to as the "type I" difference spectrum (see inset graph in figure). Some substrates cause an opposite change in spectral properties, a "reverse type I" spectrum, by processes that are as yet unclear. Inhibitors and certain substrates that bind directly to the heme iron give rise to the type II difference spectrum, with a maximum at 430 nm and a minimum at 390 nm (see inset graph in figure). If no reducing equivalents are available, this complex may remain stable, allowing the degree of binding to be determined from absorbance measurements in vitro [13] C: If carbon monoxide (CO) binds to reduced P450, the catalytic cycle is interrupted. This reaction yields the classic CO difference spectrum with a maximum at 450 nm. However, the interruptive and inhibitory effects of CO varies upon different CYPs such that the CYP3A family is relatively less affected. [15] [16]
See also
Further reading
- Estabrook RW (December 2003). "A passion for P450s (Remembrances of the early history of research on cytochrome P450)". Drug Metabolism and Disposition. 31 (12): 1461–1473. doi: 10.1124/dmd.31.12.1461. PMID 14625342. S2CID 43655270.
References
- ^ a b "Cytochrome P450". InterPro.
- ^ Danielson PB (December 2002). "The cytochrome P450 superfamily: biochemistry, evolution and drug metabolism in humans". Current Drug Metabolism. 3 (6): 561–597. doi: 10.2174/1389200023337054. PMID 12369887.
- ^ "NCBI sequence viewer". Retrieved 2007-11-19.
- ^ Nelson DR (October 2009). "The cytochrome p450 homepage". Human Genomics. 4 (1): 59–65. doi: 10.1186/1479-7364-4-1-59. PMC 3500189. PMID 19951895.
- ^ Nelson DR (January 2011). "Progress in tracing the evolutionary paths of cytochrome P450". Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 1814 (1): 14–18. doi: 10.1016/j.bbapap.2010.08.008. PMID 20736090.
- ^ Hanukoglu I (1996). "Electron transfer proteins of cytochrome P450 systems" (PDF). Adv. Mol. Cell Biol. Advances in Molecular and Cell Biology. 14: 29–55. doi: 10.1016/S1569-2558(08)60339-2. ISBN 978-0-7623-0113-3.
- ^ Tucci FJ, Rosenzweig AC (February 2024). "Direct Methane Oxidation by Copper- and Iron-Dependent Methane Monooxygenases". Chemical Reviews. 124 (3): 1288–1320. doi: 10.1021/acs.chemrev.3c00727. PMC 10923174. PMID 38305159.
- ^ [1] Archived 2019-10-18 at the Wayback Machine PROSITE consensus pattern for P450
- ^ a b Meunier B, de Visser SP, Shaik S (September 2004). "Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes". Chemical Reviews. 104 (9): 3947–3980. doi: 10.1021/cr020443g. PMID 15352783. S2CID 33927145.
- ^ Poulos TL, Finzel BC, Howard AJ (June 1987). "High-resolution crystal structure of cytochrome P450cam". Journal of Molecular Biology. 195 (3): 687–700. doi: 10.1016/0022-2836(87)90190-2. PMID 3656428.
- ^ Sligar SG, Cinti DL, Gibson GG, Schenkman JB (October 1979). "Spin state control of the hepatic cytochrome P450 redox potential". Biochemical and Biophysical Research Communications. 90 (3): 925–932. doi: 10.1016/0006-291X(79)91916-8. PMID 228675.
- ^ a b Rittle J, Green MT (November 2010). "Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics". Science. 330 (6006): 933–937. Bibcode: 2010Sci...330..933R. doi: 10.1126/science.1193478. PMID 21071661. S2CID 206528205.
- ^ a b Ortiz de Montellano PR (2005). Cytochrome P450: structure, mechanism, and biochemistry (3rd ed.). New York: Kluwer Academic/Plenum Publishers. ISBN 978-0-306-48324-0.
- ^ Huang X, Groves JT (March 2018). "Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins". Chemical Reviews. 118 (5): 2491–2553. doi: 10.1021/acs.chemrev.7b00373. PMC 5855008. PMID 29286645.
- ^ Hopper CP, Zambrana PN, Goebel U, Wollborn J (June 2021). "A brief history of carbon monoxide and its therapeutic origins". Nitric Oxide. 111–112: 45–63. doi: 10.1016/j.niox.2021.04.001. PMID 33838343. S2CID 233205099.
- ^ Smith AT, Pazicni S, Marvin KA, Stevens DJ, Paulsen KM, Burstyn JN (April 2015). "Functional divergence of heme-thiolate proteins: a classification based on spectroscopic attributes". Chemical Reviews. 115 (7): 2532–2558. doi: 10.1021/cr500056m. PMID 25763468.