An electron microscope is a
microscope that uses a beam of
electrons as a source of illumination. They use
electron optics that are analogous to the glass lenses of an optical light microscope to control the electron beam, for instance focusing them to produce magnified images or
electron diffraction patterns. As the wavelength of an electron can be up to 100,000 times smaller than that of visible light, electron microscopes have a much higher
resolution of about 0.1 nm, which compares to about 200 nm for
light microscopes.[1]Electron microscope may refer to:
Reproduction of an early electron microscope constructed by
Ernst Ruska in the 1930s
Many developments laid the groundwork of the
electron optics used in microscopes.[2] One significant step was the work of
Hertz in 1883[3] who made a cathode-ray tube with electrostatic and magnetic deflection, demonstrating manipulation of the direction of an electron beam. Others were focusing of the electrons by an axial magnetic field by
Emil Wiechert in 1899,[4] improved oxide-coated cathodes which produced more electrons by
Arthur Wehnelt in 1905[5] and the development of the electromagnetic lens in 1926 by
Hans Busch.[6] According to
Dennis Gabor, the physicist
Leó Szilárd tried in 1928 to convince him to build an electron microscope, for which Szilárd had filed a patent.[7]
To this day the issue of who invented the transmission electron microscope is controversial.[8][9][10][11] In 1928, at the
Technical University of Berlin, Adolf Matthias (Professor of High Voltage Technology and Electrical Installations) appointed
Max Knoll to lead a team of researchers to advance research on electron beams and cathode-ray oscilloscopes. The team consisted of several PhD students including
Ernst Ruska. In 1931,
Max Knoll and
Ernst Ruska[12][13] successfully generated magnified images of mesh grids placed over an anode aperture. The device, a replicate of which is shown in the figure, used two magnetic lenses to achieve higher magnifications, the first electron microscope. (Max Knoll died in 1969, so did not receive a share of the 1986
Nobel prize for the invention of electron microscopes.)
Apparently independent of this effort was work at
Siemens-Schuckert by
Reinhold Rüdenberg. According to patent law (U.S. Patent No. 2058914[14] and 2070318,[15] both filed in 1932), he is the inventor of the electron microscope, but it is not clear when he had a working instrument. He stated in a very brief article in 1932[16] that Siemens had been working on this for some years before the patents were filed in 1932, claiming that his effort was parallel to the university development. He died in 1961, so similar to Max Knoll, was not eligible for a share of the 1986 Nobel prize.[17][18]
In the following year, 1933, Ruska and Knoll built the first electron microscope that exceeded the resolution of an optical (light) microscope.[19] Four years later, in 1937, Siemens financed the work of Ernst Ruska and
Bodo von Borries, and employed
Helmut Ruska, Ernst's brother, to develop applications for the microscope, especially with biological specimens.[19][20] Also in 1937,
Manfred von Ardenne pioneered the
scanning electron microscope.[21] Siemens produced the first commercial electron microscope in 1938.[22] The first North American electron microscopes were constructed in the 1930s, at the
Washington State University by Anderson and Fitzsimmons [23] and at the
University of Toronto by
Eli Franklin Burton and students Cecil Hall,
James Hillier, and Albert Prebus. Siemens produced a transmission electron microscope (TEM) in 1939.[24] Although current transmission electron microscopes are capable of two million times magnification, as scientific instruments they remain similar but with improved optics.
In the 1940s, high-resolution electron microscopes were developed, enabling greater magnification and resolution.[25] By 1965,
Albert Crewe at the
University of Chicago introduced the scanning transmission electron microscope, enhancing imaging capabilities.[26] In the 1980s, the
field emission gun was developed for electron microscopes, improving resolution and imaging quality.
FEI Company, founded in 1971, became a major manufacturer of electron microscopes.[27] In 1991,
Tescan was established, bringing innovation with their
scanning electron microscopes (SEMs) and focused
ion beam systems.[28] The 2000s were marked by advancements in aberration-corrected electron microscopy, allowing for atomic-scale resolution.
Wavelength
Operating principle of a transmission electron microscope
The original form of the electron microscope, the
transmission electron microscope (TEM), uses a
high voltageelectron beam to illuminate the specimen and create an image. An electron beam is produced by an
electron gun, with the electrons typically at 40 to 400 keV, focused by
electromagnetic lenses, and transmitted through the specimen. When it emerges from the specimen, the electron beam carries information about the structure of the specimen that is magnified by lenses of the microscope. The spatial variation in this information (the "image") may be viewed by projecting the magnified electron image onto a
detector. For example, the image may be viewed directly by an operator using a fluorescent viewing screen coated with a
phosphor or
scintillator material such as
zinc sulfide. A high-resolution phosphor may also be coupled by means of a lens optical system or a
fibre optic light-guide to the sensor of a
digital camera.
Direct electron detectors have no scintillator and are directly exposed to the electron beam, which addresses some of the limitations of scintillator-coupled cameras.[30]
The resolution of TEMs is limited primarily by
spherical aberration, but a new generation of hardware correctors can reduce spherical aberration to increase the resolution in
high-resolution transmission electron microscopy (HRTEM) to below 0.5
angstrom (50
picometres),[31] enabling magnifications above 50 million times.[32] The ability of HRTEM to determine the positions of atoms within materials is useful for nano-technologies research and development.[33]
Transmission electron microscopes are often used in
electron diffraction mode. The advantages of electron diffraction over
X-ray crystallography are that the specimen need not be a single crystal or even a polycrystalline powder.[citation needed]
The STEM rasters a focused incident probe across a specimen. The high resolution of the TEM is thus possible in STEM. The focusing action (and aberrations) occur before the electrons hit the specimen in the STEM, but afterward in the TEM. The STEMs use of SEM-like beam rastering simplifies
annular dark-field imaging, and other analytical techniques, but also means that image data is acquired in serial rather than in parallel fashion.[citation needed]
Scanning electron microscope (SEM)
Operating principle of a scanning electron microscope
The SEM produces images by probing the specimen with a focused electron beam that is scanned across the specimen (
raster scanning). When the electron beam interacts with the specimen, it loses energy by a variety of mechanisms. The lost energy is converted into alternative forms such as heat, emission of
low-energy secondary electrons and high-energy backscattered electrons, light emission (
cathodoluminescence) or
X-ray emission, all of which provide signals carrying information about the properties of the specimen surface, such as its topography and composition.[citation needed] The image displayed by an SEM maps the varying intensity of any of these signals into the image in a position corresponding to the position of the beam on the specimen when the signal was generated. In the SEM image of an ant shown, the image was constructed from signals produced by a secondary electron detector, the normal or conventional imaging mode in most SEMs.[citation needed]
Generally, the image resolution of an SEM is lower than that of a TEM. However, because the SEM images the surface of a sample rather than its interior, the electrons do not have to travel through the sample. This reduces the need for extensive sample preparation to thin the specimen to electron transparency. The SEM also has a great depth of field, and so can produce images that are good representations of the three-dimensional surface shape of the sample.[citation needed]
In their most common configurations, electron microscopes produce images with a single brightness value per pixel, with the results usually rendered in
greyscale.[34] However, often these images are then colourized through the use of feature-detection software, or simply by hand-editing using a graphics editor. This may be done to clarify structure or for aesthetic effect and generally does not add new information about the specimen.[35]
Sample preparation for TEM
An insect
coated in gold for viewing with a scanning electron microscope
Materials to be viewed in a transmission electron microscope may require processing to produce a suitable sample. The technique required varies depending on the specimen and the analysis required:
Cryofixation – freezing a specimen so that the water forms
vitreous (non-crystalline) ice. This preserves the specimen in a snapshot of its native state. Methods to achieve this vitrification include plunge freezing rapidly in liquid
ethane, and high pressure freezing. An entire field called
cryo-electron microscopy has branched from this technique. With the development of
cryo-electron microscopy of vitreous sections (CEMOVIS)[37] and cryo-
focused ion beam milling of lamellae,[38] it is now possible to observe samples from virtually any biological specimen close to its native state.
Embedding, biological specimens – after dehydration, tissue for observation in the transmission electron microscope is embedded so it can be sectioned ready for viewing. To do this the tissue is passed through a 'transition solvent' such as
propylene oxide (epoxypropane) or
acetone and then infiltrated with an
epoxyresin such as
Araldite, Epon, or
Durcupan;[39] tissues may also be embedded directly in water-miscible
acrylic resin. After the resin has been polymerized (hardened) the sample is sectioned by
ultramicrotomy and
stained.[citation needed]
Embedding, materials – after embedding in resin, the specimen is usually ground and polished to a mirror-like finish using ultra-fine abrasives.[citation needed]
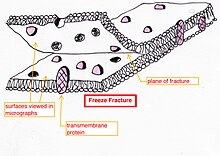
Freeze-fracture or freeze-etch – a preparation method[40][41][42] particularly useful for examining lipid membranes and their incorporated proteins in "face on" view.[43][44][45]Freeze-fracturing helps to peel open membranes to allow visualization of what is insideExternal face of bakers yeast membrane showing the small holes where proteins are fractured out, sometimes as small ring patterns.The fresh tissue or cell suspension is frozen rapidly (cryofixation), then fractured by breaking[46] (or by using a microtome)[45] while maintained at liquid nitrogen temperature. The cold fractured surface (sometimes "etched" by increasing the temperature to about −100 °C for several minutes to let some ice sublime)[45] is then shadowed with evaporated platinum or gold at an average angle of 45° in a high vacuum evaporator. The second coat of carbon, evaporated perpendicular to the average surface plane is often performed to improve the stability of the replica coating. The specimen is returned to room temperature and pressure, then the extremely fragile "pre-shadowed" metal replica of the fracture surface is released from the underlying biological material by careful chemical digestion with acids,
hypochlorite solution or
SDS detergent. The still-floating replica is thoroughly washed free from residual chemicals, carefully fished up on fine grids, dried then viewed in the TEM.[citation needed]
Freeze-fracture replica immunogold labeling (FRIL) – the freeze-fracture method has been modified to allow the identification of the components of the fracture face by immunogold labeling. Instead of removing all the underlying tissue of the thawed replica as the final step before viewing in the microscope the tissue thickness is minimized during or after the fracture process. The thin layer of tissue remains bound to the metal replica so it can be immunogold labeled with antibodies to the structures of choice. The thin layer of the original specimen on the replica with gold attached allows the identification of structures in the fracture plane.[47] There are also related methods which label the surface of etched cells[48] and other replica labeling variations.[49]
Ion beam milling – thins samples until they are transparent to electrons by firing
ions (typically
argon) at the surface from an angle and sputtering material from the surface. A subclass of this is
focused ion beam milling, where
gallium ions are used to produce an electron transparent membrane or 'lamella' in a specific region of the sample, for example through a device within a microprocessor or a
focused ion beam SEM. Ion beam milling may also be used for cross-section polishing prior to analysis of materials that are difficult to prepare using mechanical polishing.[citation needed]
Negative stain – suspensions containing
nanoparticles or fine biological material (such as viruses and bacteria) are briefly mixed with a dilute solution of an electron-opaque solution such as ammonium molybdate,
uranyl acetate (or formate), or
phosphotungstic acid.[citation needed] This mixture is applied to an EM grid, pre-coated with a plastic film such as formvar, blotted, then allowed to dry. Viewing of this preparation in the TEM should be carried out without delay for best results. The method is important in microbiology for fast but crude morphological identification, but can also be used as the basis for high-resolution 3D reconstruction using EM tomography methodology when carbon films are used for support. Negative staining is also used for observation of nanoparticles.[citation needed]
Sectioning – produces thin slices of the specimen, semitransparent to electrons. These can be cut using
ultramicrotomy on an
ultramicrotome with a glass or
diamond knife to produce ultra-thin sections about 60–90 nm thick. Disposable
glass knives are also used because they can be made in the lab and are much cheaper. Sections can also be created in situ by milling in a
focused ion beam SEM, where the section is known as a lamella.[38]
Staining – uses heavy metals such as
lead,
uranium or
tungsten to scatter imaging electrons and thus give contrast between different structures, since many (especially biological) materials are nearly "transparent" to electrons (weak phase objects). In biology, specimens can be stained "en bloc" before embedding and also later after sectioning. Typically thin sections are stained for several minutes with an aqueous or alcoholic solution of
uranyl acetate followed by aqueous lead citrate.[50]
EM workflows
Early electron microscopy of biological specimens was often descriptive, making use of the newly available higher resolution.[51] This is still the case for various applications, such as
diagnostic electron microscopy.
However, electron microscopes are now frequently used in more complex workflows, with each workflow typically using multiple technologies to enable more complex and/or more quantitative analyses of a sample. A few examples are outlined below, but this should not be considered an exhaustive list. The choice of workflow will be highly dependent on the application and the requirements of the corresponding scientific questions, such as resolution, volume, nature of the target molecule, etc.
For example, images from light and electron microscopy of the same region of a sample can be overlaid to correlate the data from the two modalities. This is commonly used to provide higher resolution contextual EM information about a fluorescently labelled structure. This correlative light and electron microscopy (
CLEM)[52] is one of a range of correlative workflows now available. Another example is high resolution mass spectrometry (ion microscopy), which has been used to provide correlative information about subcellular antibiotic localisation,[53] data that would be difficult to obtain by other means.[citation needed]
The initial role of electron microscopes in imaging two-dimensional slices (TEM) or a specimen surface (SEM with secondary electrons) has also increasingly expanded into the depth of samples.[54] An early example of these ‘
volume EM’ workflows was simply to stack TEM images of serial sections cut through a sample. The next development was virtual reconstruction of a thick section (200-500 nm) volume by backprojection of a set of images taken at different tilt angles -
TEM tomography.[55]
Serial imaging for volume EM
To acquire
volume EM datasets of larger depths than TEM tomography (micrometers or millimeters in the z axis), a series of images taken through the sample depth can be used. For example, ribbons of serial sections can be imaged in a TEM as described above, and when thicker sections are used, serial TEM tomography can be used to increase the z-resolution. More recently, back scattered electron (BSE) images can be acquired of a larger series of sections collected on silicon wafers, known as SEM array tomography.[56][57] An alternative approach is to use BSE SEM to image the block surface instead of the section, after each section has been removed. By this method, an ultramicrotome installed in an SEM chamber can increase automation of the workflow; the specimen block is loaded in the chamber and the system programmed to continuously cut and image through the sample. This is known as serial block face SEM.[58] A related method uses
focused ion beam milling instead of an ultramicrotome to remove sections. In these serial imaging methods, the output is essentially a sequence of images through a specimen block that can be digitally aligned in sequence and thus reconstructed into a
volume EM dataset. The increased volume available in these methods has expanded the capability of electron microscopy to address new questions,[54] such as mapping neural connectivity in the brain,[59] and membrane contact sites between organelles.[60]
Disadvantages
JEOL transmission and scanning electron microscope made in the mid-1970s
Electron microscopes are expensive to build and maintain. Microscopes designed to achieve high resolutions must be housed in stable buildings (sometimes underground) with special services such as magnetic field canceling systems.[61]
The samples largely have to be viewed in
vacuum, as the molecules that make up air would scatter the electrons. An exception is
liquid-phase electron microscopy[62] using either a closed liquid cell or an environmental chamber, for example, in the
environmental scanning electron microscope, which allows hydrated samples to be viewed in a low-pressure (up to 20
Torr or 2.7 kPa) wet environment. Various techniques for
in situ electron microscopy of gaseous samples have been developed.[63]
Scanning electron microscopes operating in conventional high-vacuum mode usually image conductive specimens; therefore non-conductive materials require conductive coating (gold/palladium alloy, carbon, osmium, etc.). The low-voltage mode of modern microscopes makes possible the observation of non-conductive specimens without coating. Non-conductive materials can be imaged also by a variable pressure (or environmental) scanning electron microscope.[citation needed]
Small, stable specimens such as
carbon nanotubes,
diatom frustules and small mineral crystals (asbestos fibres, for example) require no special treatment before being examined in the electron microscope. Samples of hydrated materials, including almost all biological specimens, have to be prepared in various ways to stabilize them, reduce their thickness (ultrathin sectioning) and increase their electron optical contrast (staining). These processes may result in artifacts, but these can usually be identified by comparing the results obtained by using radically different specimen preparation methods. Since the 1980s, analysis of
cryofixed, vitrified specimens has also become increasingly used by scientists, further confirming the validity of this technique.[64][65][66]
^"The Scale of Things". Office of Basic Energy Sciences, U.S. Department of Energy. 2006-05-26. Archived from
the original on 2010-02-01. Retrieved 2010-01-31.
^Luft, J.H. (1961). "Improvements in epoxy resin embedding methods". The Journal of Biophysical and Biochemical Cytology. Vol. 9, no. 2. p. 409.
PMC2224998.
PMID13764136.
^Meryman H.T. and Kafig E. (1955). The study of frozen specimens, ice crystals and ices crystal growth by electron microscopy. Naval Med. Res. Ints. Rept NM 000 018.01.09 Vol. 13 pp 529–544
^Rash, J. E.; Johnson, T. J.; Hudson, C. S.; Giddings, F. D.; Graham, W. F.; Eldefrawi, M. E. (1982-11-01). "Labelled-replica techniques: post-shadow labelling of intramembrane particles in freeze-fracture replicas". Journal of Microscopy. 128 (Pt 2): 121–138.
doi:
10.1111/j.1365-2818.1982.tb00444.x.
PMID6184475.
S2CID45238172.
^Abbott LF, Bock DD, Callaway EM, Denk W, Dulac C, Fairhall AL, Fiete I, Harris KM, Helmstaedter M, Jain V, Kasthuri N, LeCun Y, Lichtman JW, Littlewood PB, Luo L, Maunsell JH, Reid RC, Rosen BR, Rubin GM, Sejnowski TJ, Seung HS, Svoboda K, Tank DW, Tsao D, Van Essen DC (September 2020).
"The Mind of a Mouse". Cell. 182 (6): 1372–1376.
doi:10.1016/j.cell.2020.08.010.
PMID32946777.
S2CID221766693.
^Gai, P.L.; Boyes, E.D. (2009). "Advances in atomic resolution in situ environmental transmission electron microscopy and 1A aberration corrected in situ electron microscopy". Microsc Res Tech. 72 (3): 153–164.
arXiv:1705.05754.
doi:
10.1002/jemt.20668.
PMID19140163.
S2CID1746538.